The European Council has agreed to extend the deadline for the certification of medical devices in order to mitigate the risk of device shortages. The required dates are now staggered based on the device risk classification. In addition, removal of the “sell-off” date requirement will further address the concern of shortages.
These changes don’t impact the requirements of the original regulations, they just provide more time to implement them. Manufacturers now have until 2027 or 2028, depending on risk classification, to get their devices designated under MDR.
Companies selling medical devices in the European Union (EU) will soon need to meet a new – and complex – set of regulations.
The new EU MDR regulation is four-times longer than the previous regulations, and will greatly affect what information goes onto product labels and packaging.
MDR presents both a data and design dilemma, as the labeling of medical devices will come under greater scrutiny in the name of patient safety. Brands selling products in the EU must meet these new regulations if they want to stay in the market.
What You Need to Know About MDR
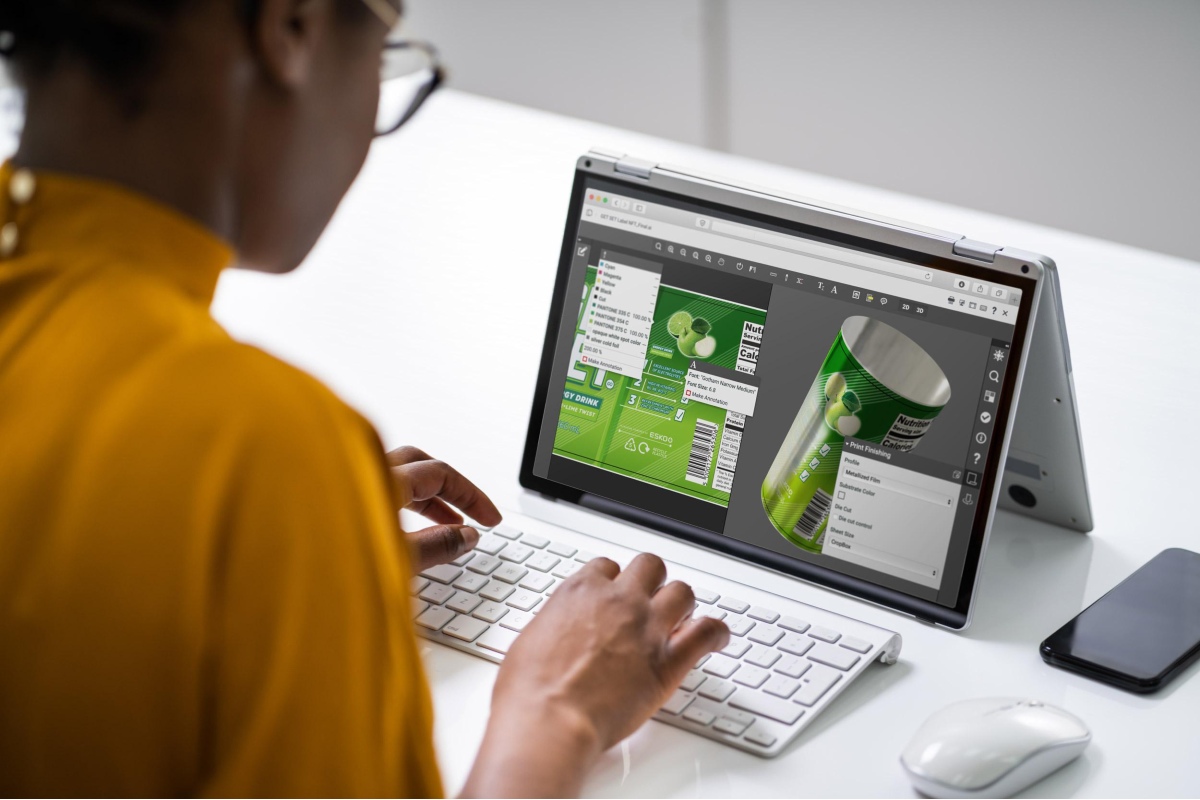
MDR requires manufacturers to display a new set of information and symbols on product labels. Specifically, labels will need to show unique device identification (UDI) numbers—used to track devices—and instructions for use (IFU).
IFUs will also be required on product information channels, like a company’s website. IFUs must allow patients to see what the use of the device is, guidelines for reuse, warnings related to device disposal, and incident reports in relation to the device.
MDR states that medical device labeling must be in the national language and “clearly comprehensible to the intended user or patient.” Since people speak more than 30 languages in the EU, a number of symbols have been developed to make labels as universally comprehensible as possible:
- A crossed-out box for “do not use if the package is damaged”
- An umbrella with raindrops for “keep this package dry”
- A box containing the letters “UDI” to be placed adjacent to the UDI number
Under the new regulations, importers must print their name on the IFU, device, or packaging, and companies that translate, re-label, or re-package devices will also need to place their name on the product.
Ready for more? MDR won’t just affect new products. Existing medical devices will need to be recertified, and the definition of “medical device” has changed to include products such as contact lenses and cleaning products for certain devices. Translation: a lot of SKUs are about to get a packaging makeover.
What Pharma Brands Can Do to Prepare
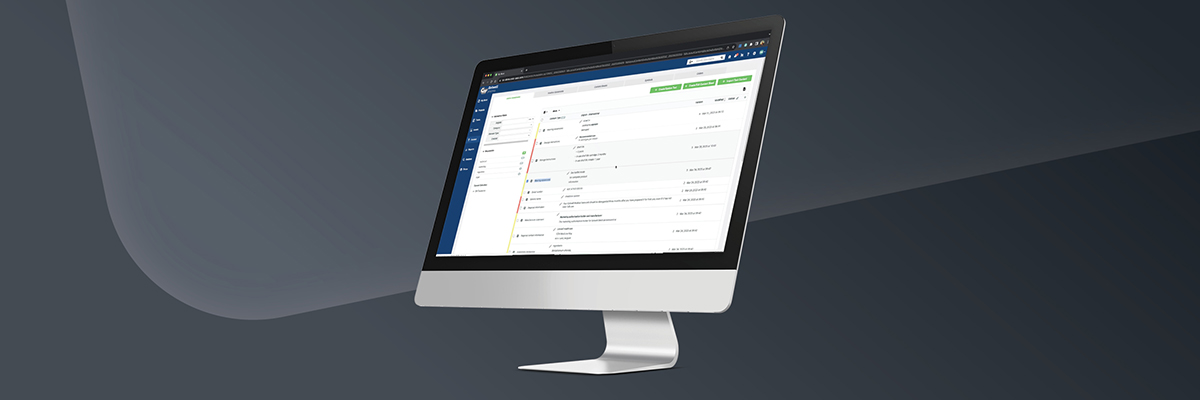
With more than 30 languages and myriad regulations to track, it’ll be no easy task for medical device companies to adhere to the new MDR regulations. However, one way to reduce the mayhem is to adopt a packaging management system to track all of the necessary packaging changes – and the workflows involved in each – from design brief to production.
A good label and artwork management system will allow Medical Device companies to simplify the packaging content management process for label changes by giving brands a central repository for storing all essential data related to each medical device, with set permissions for who can view, download, and edit files. You can also track version changes, and so can outside parties such as clients, agencies, and printers.
Brands that use a label and artwork management system can also create new templates and automatically populate the templates with approved data from the repository, so packaging designers have a realistic view of the volume and types of data elements they’re organizing on the new package.
In addition, workflow management features allow all stakeholders (design, translation, regulatory agencies, printer converters, and more) to simultaneously view and collaborate on projects in progress, which improves quality control and simplifies the packaging approval process – making sure each new package is “shelf-ready” by the new deadlines.